Nueva variante genética del gen HPS1 en el síndrome de Hermansky-Pudlak con progresión fulminante de fibrosis pulmonar: un informe de caso
Fondo
El síndrome de Hermansky-Pudlak (HPS) es un trastorno autosómico recesivo asociado con albinismo oculocutáneo (o algún grado de hipopigmentación), disminución de la actividad visual generalmente acompañada de nistagmo horizontal, diátesis hemorrágica, colitis granulomatosa y fibrosis pulmonar altamente penetrante en algunos subtipos. El espectro de HPS incluye diez trastornos (HPS-1 a HPS-10). Las mutaciones homocigotas o heterocigotas compuestas en HPS1, HPS3, HPS4 y varios otros genes conducen a la manifestación clínica de la enfermedad [1,2,3]. La enfermedad fue descrita por dos hematólogos checos František Heřmanský y Pavel Pudlák en 1959 [4].
Presentación del caso
Mujer caucásica de 57 años (proband), profesora, con albinismo oculocutáneo (Fig. 1)ingresó por tos seca y rápido empeoramiento de la disnea. Un análisis exhaustivo de la historia clínica reveló que la paciente tenía problemas oculares desde la infancia y que desde los 45 años, su visión era significativamente peor. Además, se encontró que tuvo varios episodios de sangrado prolongado: después de la apendicectomía, después de lesiones menores (incluidos los hemartros) y después del parto. A la edad de 50 años, fue examinada por un hematólogo. Se realizó agregación plaquetaria, mostrando un tiempo ligeramente prolongado de PFA-100 en presencia de colágeno/ADP. No se ha llegado a una conclusión definitiva con respecto a este hallazgo. Los primeros problemas pulmonares leves ocurrieron a la edad de 53 años. Fue seguida con diagnóstico de asma bronquial por un neumólogo regional. No había antecedentes familiares de estos síntomas. Era una no fumadora.
Figura 1

Albinismo en el paciente con síndrome de Hermansky-Pudlak
El examen físico reveló dedos a garroteos y crepitaciones finas inspiratorias finales bilaterales en las áreas pulmonares inferior y media. La radiografía de tórax posteroanterior mostró opacidades reticulares difusas bilaterales.
La tomografía computarizada (CTARR) de alta resolución del tórax identificó engrosamiento del intersticio peribroncovascular, bronquiectasias, reticulaciones, panal, opacidades de vidrio esmerilado y consolidaciones del parénquima pulmonar. Una comparación de las imágenes de HRCT realizadas a intervalos de 3 meses mostró una progresión fulminante de la afectación pulmonar (Figs. 2 y 3).
Figura 2

Tomografía computarizada de alta resolución (plano axial) del tórax que muestra empeoramiento de la fibrosis pulmonar con engrosamiento del intersticio peribroncovascular, bronquiectasias, reticulaciones, panal, opacidades de vidrio esmerilado y consolidaciones del parénquima pulmonar. Examen inicial(a , b) y seguimiento de tres meses (c, d)
Fig. 3

Tomografía computarizada de alta resolución (plano sagital) del tórax que muestra empeoramiento de la fibrosis pulmonar con engrosamiento del intersticio peribroncovascular, bronquiectasias, reticulaciones, panal, opacidades de vidrio esmerilado y consolidaciones del parénquima pulmonar. Examen inicial(a ) y seguimiento de tres meses (b)
Las pruebas de función pulmonar revelaron un deterioro severo de la ventilación restrictiva y una disminución severa de la capacidad de difusión del pulmón para el monóxido de carbono (DLco 20%). El análisis de gasodemia arterial mostró hipoxemia (p02 7 kPa). Se encontró hipertensión pulmonar moderada. El hemograma, la bioquímica sérica y los parámetros inmunológicos fueron normales.
Sobre la base de estos hallazgos, se sospechó HPS. Los antecedentes familiares negativos de los síntomas sugirieron un modo de herencia autosómico-recesivo. Por lo tanto, se llevó a cabo la secuenciación completa del exoma (una forma de secuenciación paralela masiva) del trío proband-parent. Se recolectaron muestras de sangre periférica y se procesaron para el aislamiento del ADN genómico utilizando MagCore® Genomic DNA Whole Blood Kit (RBC Bioscience). Las bibliotecas completas de exomas se procesaron utilizando KAPA Hyper Prep Kit, SeqCap EZ MedExome Enrichment Kit y HyperCap Bead Kit (Roche, EE. UU.) de acuerdo con SeqCap EZ HyperCap Workflow v2.1 siguiendo los protocolos recomendados. Se realizó una secuenciación de 2 × 75 pb en el secuenciador Illumina NextSeq 500 (Illumina Inc., EE. UU.). Las lecturas de secuenciación en bruto se alinearon con el genoma de referencia humano GRCh37 (hg19) utilizando el algoritmo BWA-mem, versión 0.7.15, los duplicados de PCR se identificaron con la herramienta MarkDuplicates de Picard. Las variantes de nucleótido único de la línea germinal (SNV) y los indels fueron detectados por el GATK HaplotypeCaller, versión 3.7. La anotación de las variantes/indels obtenidas se realizó con Annovar. Además, las variantes /indels procesadas se emparejaron con el panel virtual de genes que incluyen HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3 y PLDN. El panel virtual fue creado a partir de la revisión de la literatura [1,2,3]. La secuenciación del exoma identificó un genotipo heterocigoto compuesto en el gen HPS1 (NM_000195.3) en la probanda: 1) variante patógena de desplazamiento de marco c.1189delC; p.(Gln397Serfs*2), lo que resulta en un codón de parada prematuro, asociado con HPS; y 2) variante sin sentido previamente no descrita, c.1507C > T; p.(Gln503*), lo que resulta en un codón de parada prematuro, lo que implica una pérdida de 197 aminoácidos o, más probablemente, una desintegración mediada por tonterías de la degradación del ARNm (Fig. 4).
Figura 4

Visualización de la variante c.1507C > T (g.100183535C > T)(a ) y de la variante c.1189delC (g.100185444) (b) por Intergrative Genomics Viewer. Las variantes están marcadas por marcos rojos. Las lecturas de secuenciación hacia adelante están en azul; las lecturas de secuenciación inversa están en rosa
El rango de cobertura de la nueva variante c.1507C > T en la probanda fue de 26 lecturas y el rango de frecuencia del alelo variante 38,5%.
Posteriormente, se ha verificado el diagnóstico mediante PCR y secuenciación de Sanger de los amplicones en la probanda, así como la presencia de estado portador heterocigoto de los padres (Fig. 5). Los cebadores fueron diseñados para los exones 13 y 15, respectivamente (13F-primer: CTTAGGGTTGGCACGTCTTC, 13R-primer: TGGGTCTCACCTGAATCTCC; 15F-primer: TTCTGCTGTAATGCCCTCCT, 15R-primer: GAAGTCCTTCCAGTCCGTCA). La PCR se realizó con la temperatura de recocido de 60 °C utilizando la ADN polimerasa de alta fidelidad Q5 (New England Biolabs Inc., Inglaterra) de acuerdo con el protocolo del fabricante. Los productos de PCR se purificaron utilizando el kit de purificación Qiaquick PCR (QIAGEN, Alemania). La secuenciación capilar se realizó utilizando la química BigDye-terminator en 3500 Genetic Analyzer (Applied Biosystems, EE. UU.).
Figura 5
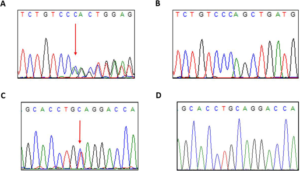
Resultados de la secuenciación de Sanger de la variante c.1189delC (a) en comparación con la variante de tipo salvaje (b) y de la variante c.1507C > T (c) en comparación con la variante de tipo salvaje (d ))
No fue posible analizar el efecto estructural de la variante p.(Gln503*) “in silico” porque la estructura cristalina no estaba disponible. Sin embargo, dado el tipo de variante “sin sentido”, podemos suponer que la nueva variante que incluye el cambio de nucleótido C > T en la posición 1507 conduce a una proteína acortada que muy probablemente resulta en un mal plegamiento de la proteína y deterioro de la función.
El tratamiento con corticosteroides, iniciado antes de que se confirmara el diagnóstico de HPS, no tuvo ningún efecto sobre las funciones pulmonares. Por lo tanto, el trasplante de pulmón comenzó a prepararse. Desafortunadamente, 2 meses después del diagnóstico de HPS, el paciente murió debido a la fibrotización pulmonar fulminante en curso.
Discusión y conclusiones
El HPS es una enfermedad autosómica recesiva rara y heterogénea caracterizada por anomalías tanto en los lisosomas como en los orgánulos relacionados con los lisosomas. La enfermedad es rara en caucásicos, pero es la causa más prevalente de albinismo en Puerto Rico [5]. Se han notificado diez subtipos de HPS (HPS-1 a HPS-10); tres subtipos de HPS están asociados con la enfermedad pulmonar fibrótica: HPS-1, HPS-2 y HPS-4. HPS puede ser causada por al menos nueve genes: HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3 y PLDN.
Hasta la fecha, se han reportado 61 variantes en el gen HPS1 como causantes de enfermedades o probablemente causantes de enfermedades de acuerdo con la Base de Datos de Mutaciones genéticas humanas (Tabla 1)[7]. Las variantes patogénicas más comunes del gen HPS1 son tonterías/sentidos insensales o pequeñas deleciones. La fibrosis pulmonar se observa en aproximadamente la mitad de los portadores de mutaciones de los genes HPS1 y HPS4 [2, 3, 6]. Sin embargo, otras variantes del gen HPS1 se asocian con síntomas más leves como albinismo, nistagmo, hipopigmentación, hipoplasia foveal o ausencia de uñas [7].
Se ha descrito que la mayoría de los pacientes con HPS son heterocigotos compuestos [8]. Nuestra probanda también fue un heterocigoto compuesto portador de la variante de desplazamiento de marco previamente descrita c.1189delC y la nueva variante sin sentido c.1507C > T. Theunissen et al. informaron un paciente que era heterocigoto compuesto con la misma variante c.1189delC que en nuestro caso y diferente variante sin sentido c.517C > T. Este paciente padecía un albinismo oculocutáneo y una “enfermedad multisistémica” desde la infancia [9]. Hermos et al. describieron cuatro nuevas variantes de HPS1 en pacientes no puertorriqueños que sufrían de HPS, donde se han encontrado pequeñas deleciones del nucleótido C (c.561delC) y el nucleótido A (c.1581delA) del gen HPS1 que no producen ARN. Uno de estos pacientes desarrolló fibrosis pulmonar, dos pacientes tenían colitis granulomatosa [10]. Hasta ahora se han descrito ocho variantes causantes de enfermedades del tipo sin sentido. En una familia pakistaní, una variante sin sentido p.(Gln686*) del gen HPS1 se segregaba con el fenotipo HPS. La ausencia de fibrosis pulmonar en estos individuos afectados podría deberse a su edad relativamente joven [11]. Por otro lado, Abouelhoda et al. detectaron una variante hpS1 sin sentido en el exón 14 asociada solo con uñas ausentes [12].
Los subtipos de HPS con fibrosis pulmonar tienen un peor pronóstico en comparación con otros tipos de HPS. Las manifestaciones clínicas de la fibrosis pulmonar asociada a HPS ocurren generalmente en la cuarta o quinta década de la vida [1,2,3].
Los hallazgos radiológicos de la fibrosis pulmonar HPS son variables: opacidades reticulares, engrosamiento septal y pleural, bronquiectasias, opacidades de vidrio esmerilado, pérdida de volumen pulmonar o panal. Los hallazgos radiográficos predominantes se encuentran en la periferia pulmonar y el progreso hacia la porción central del pulmón [13].
La esperanza de vida promedio de los pacientes con HPS es de 40 a 50 años. La fibrosis pulmonar es una causa común de muerte en pacientes con HPS [13, 14]. No existe una terapia curativa conocida para hps. Los corticosteroides no son efectivos. Se ha demostrado que la pirfenidona, un agente antifibrótico, retrasa la progresión de la fibrosis, pero solo en pacientes que tienen un volumen pulmonar residual bien conservado [3, 7]. Por lo tanto, el trasplante de pulmón sigue siendo el único medio de prolongar la supervivencia de los pacientes con HPS con fibrosis pulmonar avanzada [15]. Una contraindicación potencial para realizar un trasplante de pulmón es la trombocitopatía asociada con HPS. Esta condición puede ser manejada por la administración intravenosa de desmopresina y transfusiones de plaquetas [16].
Las mutaciones heterocigotas compuestas en HPS1 en nuestra proband condujeron a la interrupción del gen HPS1 y a la manifestación clínica de HPS con fibrosis pulmonar grave. Este caso ilustra la necesidad de considerar la HPS en el diagnóstico diferencial de la fibrosis pulmonar. El diagnóstico temprano de HPS puede ayudar al momento del trasplante de pulmón. Nuestro caso también muestra que la progresión de la fibrosis asociada a HPS puede ser fulminante. Por lo tanto, una indicación para el trasplante de pulmón no se puede retrasar.
Abreviaturas
DLco: Capacidad difusora del pulmón para el monóxido de carbono
HPS: Síndrome de Hermansky-Pudlak
HRCT: Tomografía computarizada de alta resolución
NGS: Secuenciación de próxima generación
SNV: Variantes de un solo nucleótido
Recopilado y traducido al español desde www.bmcpulmmed.biomedcentral.com
(Publicado originalmente el 16 de Octubre del 2021)CITA: Doubková, M., Trizuljak, J., Vrzalová, Z. et al. Novel genetic variant of HPS1 gene in Hermansky-Pudlak syndrome with fulminant progression of pulmonary fibrosis: a case report. BMC Pulm Med19, 178 (2019). https://doi.org/10.1186/s12890-019-0941-4
DOI: https://doi.org/10.1186/s12890-019-0941-4